News and Publications

Mankind Pharma and Denovo Sciences
join forces to drive AI-led innovation in drug discovery
Mankind Pharma has announced a strategic collaboration with Denovo Sciences to launch an AI-led drug discovery programme, marking an important step in the company’s focus on innovation-driven and technology-enabled research and development.

Multi-target computational pipeline for discovery of pan-influenza neuraminidase
In this study, we developed a multi-target computational pipeline to discover broad-spectrum influenza neuraminidase inhibitors. By screening nearly 500,000 compounds and applying cross-validation across influenza A and B neuraminidase subtypes, we identified promising candidates with stable pan-influenza binding profiles.

Data-driven discovery of chemical signatures for developing new inhibitors against human influenza viruses
In this study, we mapped the antiviral chemical space of influenza A and B by curating over 400,000 molecules from public databases. We identified key chemical signatures, promising scaffolds, and multi-target candidates that can guide the design of next-generation influenza inhibitors.

Discovery of new antiviral agents through artificial intelligence-based de novo design targeting influenza virus neuraminidase
In this foundational paper, we showed that the Denovo Platform can generate optimized small molecules with potent anti-influenza activity in both in vitro and in vivo studies. The work provides direct proof that our AI-driven approach can move from computational design to experimentally validated antiviral candidates.

Computational evaluation and benchmark study of 342 crystallographic holo-structures of SARS-CoV-2 Mpro enzyme
In this study, we systematically benchmarked 342 SARS-CoV-2 Mpro holo-structures and showed that even closely related PDB conformations can perform very differently in virtual screening and pose prediction. This work provides a strong scientific foundation for the importance of Q-pocket, which was developed to identify the most suitable target conformations for structure-based drug discovery.

Targeting SARS-CoV-2 main protease: a comprehensive approach using advanced virtual screening, molecular dynamics, and in vitro validation
In this study, we combined pharmacophore modeling, advanced virtual screening, molecular dynamics, and in vitro assays to identify new SARS-CoV-2 main protease inhibitors. From screening around 200 million compounds, we discovered two micromolar hits that provide promising scaffolds for further antiviral optimization.

Can artificial intelligence transform antiviral drug discovery?
In this review, we explore how AI is beginning to reshape antiviral drug discovery, from target identification and drug repurposing to de novo molecular design and resistance prediction. We also discuss the key limitations and challenges that must be addressed for AI-driven antivirals to achieve real translational impact.

Machine Learning-Based Soft Voting Ensemble Model for the Prediction of Oral Drug-Likeness of Chemical Structures
HADES is a machine learning tool that predicts drug-likeness by integrating multiple molecular properties, helping researchers prioritize promising compounds e…HADES is a machine learning tool that predicts drug-likeness by integrating multiple molecular properties, helping researchers prioritize promising compounds early in drug discovery. It outperforms existing models in distinguishing approved drugs from non-drugs and identifies problematic structures. The tool effectively guides molecular optimization and enriches virtual screening results, making it a practical resource for compound prioritization in drug discovery.
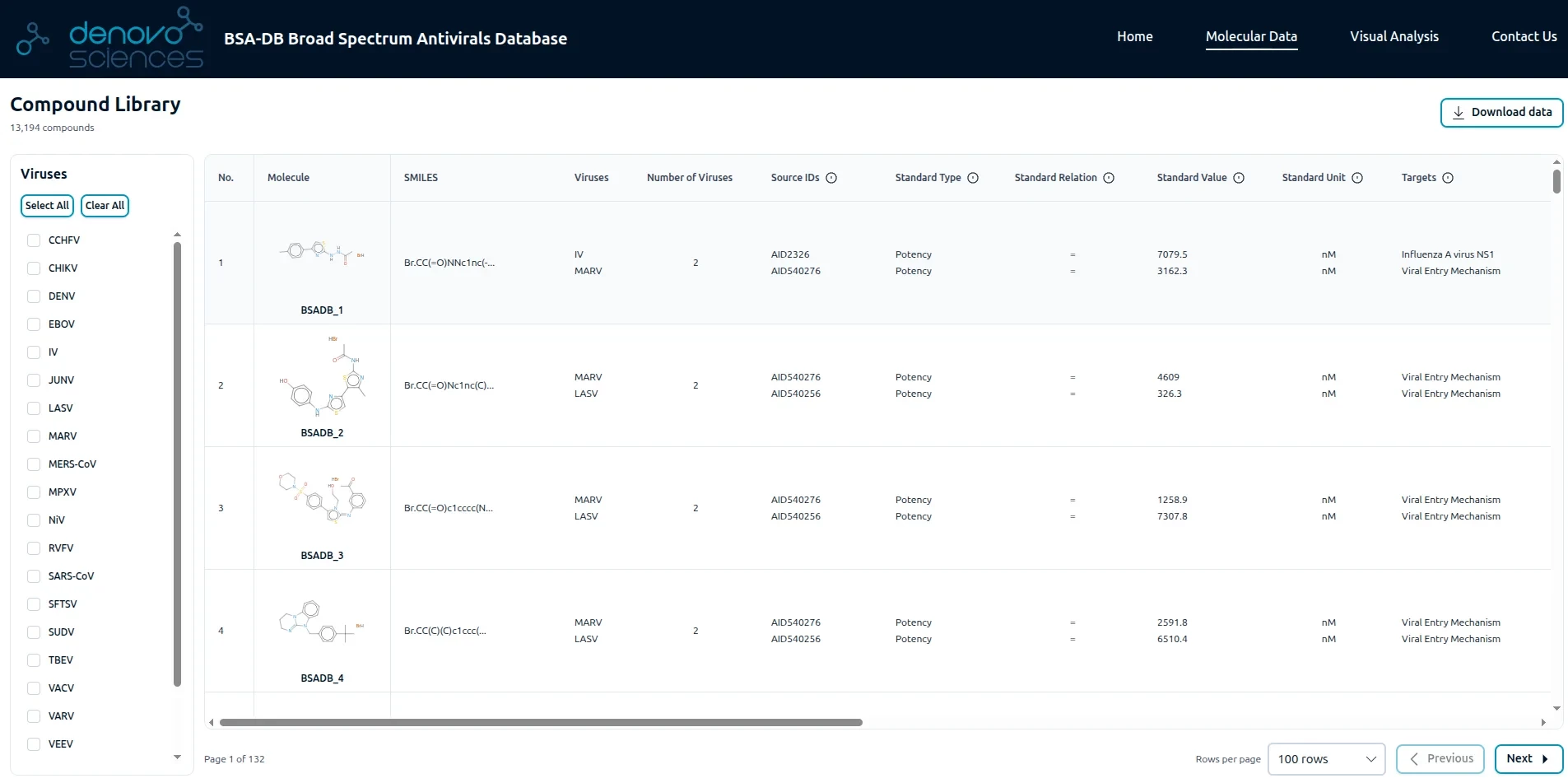
Broad-Spectrum Antiviral Database (BSAD)
This project presents a comprehensive, curated database of broad-spectrum antiviral compounds effective against high-priority pandemic pathogens identified by the WHO. The database, compiled from three major sources (PubChem, BindingDB, and ChEMBL),includes molecules with activity against multiple viruses at concentrations below 10,000 nM. These broad-spectrum antivirals are valuable for rapid therapeutic development during emerging outbreaks and pandemic preparedness.
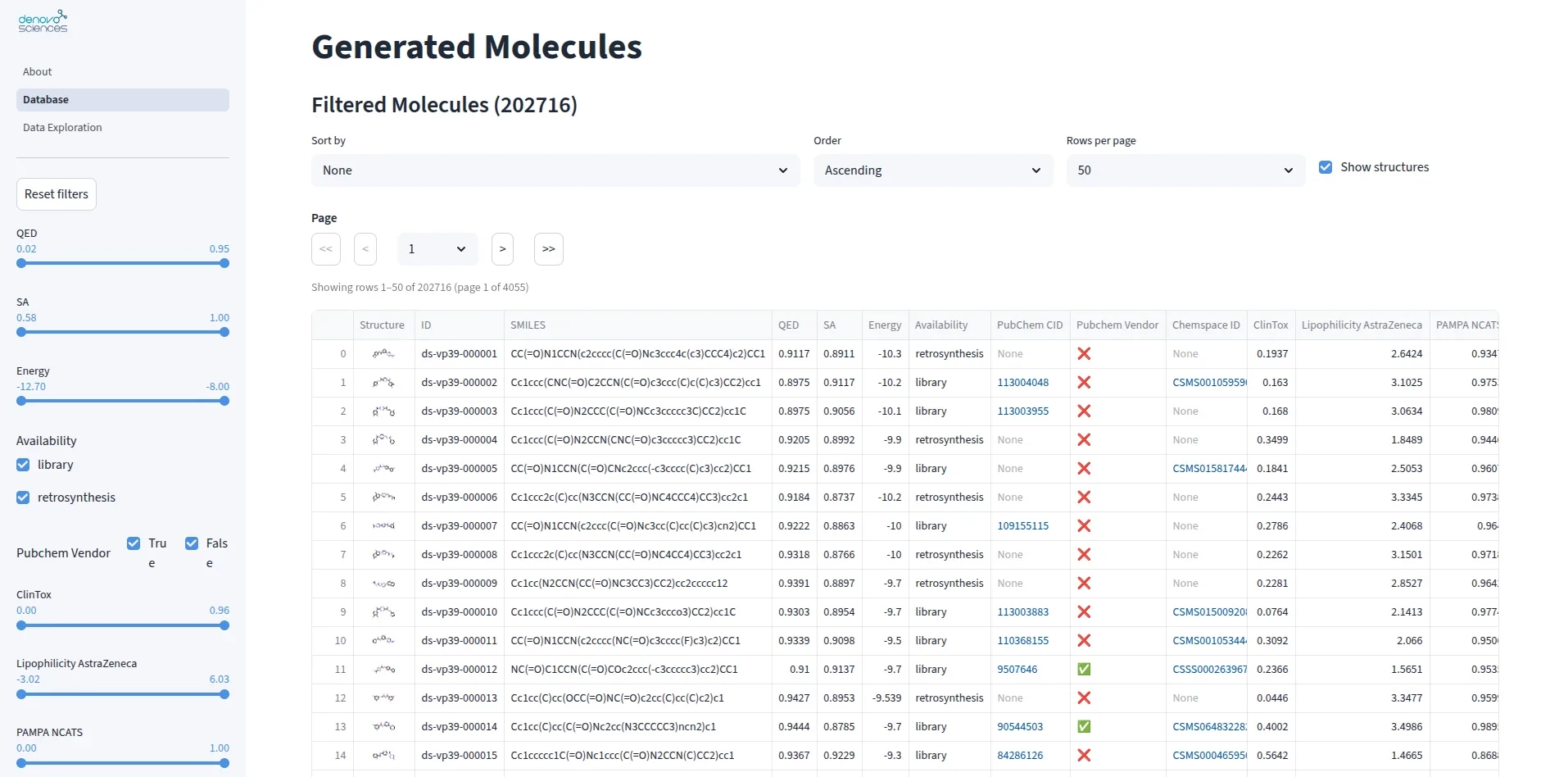
Database of de novo designed small molecules targeting the Mpox virus
Mpox virus is an emerging health threat with limited treatment options. Mpox virus is an emerging health threat with limited treatment options. We created a dataset of over 202,000 small molecules designed to target VP39, a key viral protein, using artificial intelligence optimized for binding effectiveness and drug-likeness. Each molecule includes detailed annotations on binding predictions, chemical properties, toxicity, and synthesis feasibility. This annotated library accelerates drug discovery by providing a starting point for identifying and testing promising Mpox treatments.